+86 13683842418
+86 13683842418
河南省洛阳市洛阳国家大学科技园
摘要
宏基因组学研究中,因不同技术产生的差异必须最小化,以准确评价微生物对人类健康的影响。为此,作者使用21种DNA提取方法,对同一粪便样本的微生物群落差异进行了量化分析。作者发现,DNA的提取方法对宏基因组学结果有很大的影响。为了将DNA提取方法进行分级,作者分析了所得DNA的质量与数量,并对菌群多样性及革兰氏阴性菌与革兰氏阳性菌的比率进行了评估。在使用了已知成分的菌群模型进行校对后,作者推荐了一种可以通用于各实验室的标准粪便DNA提取方法。这一标准方法的使用,将会提高人类肠道菌群研究的可比较性,并促进meta分析的发展。
前言
近5年来,有超过3000篇的学术论文使用DNA或RNA的基础分析方法对人体中的微生物群落进行研究,目前文献中有160,000个宏基因组。人类消化道系统是研究最多的一个。肠道微生物因其数量及多样性且与人类健康和疾病息息相关而备受关注。许多研究根据特殊的微生物图谱,来判断如糖尿病、炎症性肠病和结肠直肠癌等疾病的状态,但这些研究几乎都各自使用不同的技术和统计方法,限制了meta分析研究。
宏基因组学的研究中,每一步技术的变化而变化。样本获得的部位、存储方式等都会影响测定的结果,因此在一些研究中大的样本在保存前应进行均质化,获得多个完全相同的小份,-20℃以下保存为常规选择。
作者研究了DNA的提取方法对所得到的微生物群落组成结果的影响程度,并进行了量化处理。本实验作者应用了严格且一致的质量标准,系统地研究了同一方法在同一实验室和不同实验室(三个大洲)的可重复性。之后,作者使用由已知相对丰度的10种微生物组成模拟菌群(同时包含革兰氏阳性菌和革兰氏阴性菌),对评分最好的DNA分析方法的精准性进行了检验。最终,作者推荐了一个提取人类粪便样本DNA的标准方法。如果这一方法被广大研究者所接受并使用,一定会大大提高宏基因组学研究的可比性。
结果
研究设计
研究分为两个阶段。第一阶段,评估不同DNA提取方法所带来的差异。作者将两例粪便样本分成多个小份(样本来源于两个个体,分别命名为样本A和样本B),然后分成200mg的小份置于-80℃冰冻4h。每个样本取出4份儿,干冰运输至3个洲的11个国家的21个合作实验室。参与的实验室共使用7种常见的试剂盒和1种非试剂盒法来提取样本DNA。每个实验室至少使用4种提取方法,随后将提取的DNA运送至同一个测序中心(GENOSCOPE, France)进行测序。在第二阶段,综合考虑了所提取的DNA的数量、完整性、多样性和革兰氏阳性菌的比率后,作者选择了五个方法,将其中十分相似的三种方法合并为一种方法,最后用三种方法进行比较。每种方法由最初使用的实验室和另外三个从没接触过该方法的实验室共同执行,以评估该方法在实验室之间的可重复性。为了量化3种提取方法的绝对误差,作者准备了由10种细菌构成的模拟菌群,进一步研究方法的可重复性。
DNA产量和断裂程度的质量控制
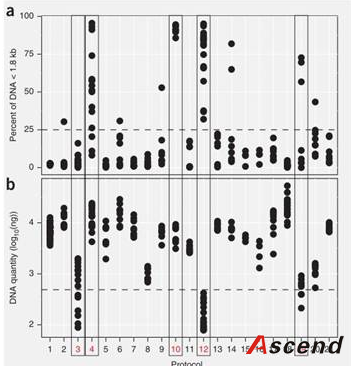
提取产量的最大化和片段最小断裂程度是选择一个DNA提取方法的关键,过去的研究结果相同,不同方法提取的DNA的量有相当大的变化(fig. 2)。如方法18所获得的DNA数量是方法3和12的100倍,是方法8、19和20的10倍。此外,所获得的DNA的断裂情况也有很大的不同,以长度在1.8 kb以下的DNA占总DNA的百分比进行比较;方法4、10和12产生高片段化的DNA,而方法1没有片段化的DNA。随后的分析发现,DNA获得量低于500 ng或DNA片段化程度过高(中位样本破碎度25%以上)的样本不能用于测序。
菌群变异性质量控制
以IlluminaHiSeq2000对提取到的DNA进行鸟枪法测序,使用MOCAT29pipeline进行处理,计算相对的种类丰度和基因功能丰度,随后用人类肠道菌群的内参基因进行分析。
作者发现不同的样本处理方法和批次影响了分析的结果,尽可能的减小这种偏差十分重要。最大的差异来自于个体间,其次差异来自于个体内部,但这一差别远小于个体之间的差别,证明了每个个体有自己独特的肠道菌群。作者使用相关分析来评估物种的丰度,发现提取方法间的差异要小于同一技术提取不同样本间的差异(Fig. 3a)。基于直系同源组分析发现,提取方法的影响大于样本内部的差异和同一个体不同时间样本的差异(Fig. 3b),储存方法不同造成的影响大于方法内产生的影响并小于样本间的差异。
总的来说,这些结果表明不同的DNA提取方法,对测量群落的组成和功能产生了影响。

物种丰度质量控制
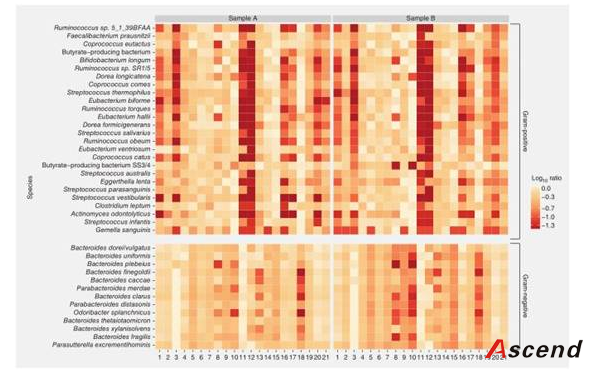
作者调查了物种特异性和丰度变化,来估计哪一个受提取方法影响最大。由于高机械强度的细胞壁的差异,革兰氏阳性菌更容易受提取方法的影响。大多数方法明显缺少了革兰氏阳性菌的部分,而对革兰氏阴性菌的影响很小(Fig. 4)。使用Shannon多样性分析,发现所得到的多样性与革兰氏阳性菌的回收丰度有关,革兰氏阳性菌的比例越高,多样性越高(补充Fig. 3)。因此得到结论,多样性是判断方法的性能和获得DNA丰度的一个很好指标。
影响DNA提取结果的因素
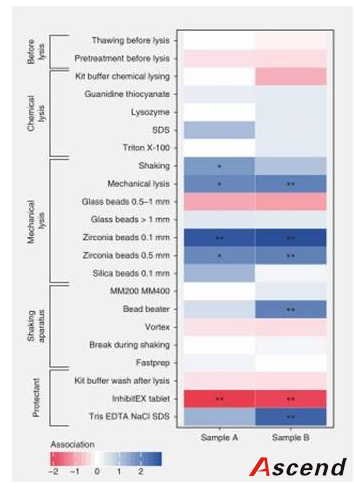
将菌群多样性作为最优准则,作者确定了方法的相关因素(Fig. 5)。发现机械裂解、氧化锆珠和震荡的使用与所获得的菌群多样性呈正相关。唯一负相关的是InhibitEX tablet(Fig.5),研究表明,这一试剂对DNA的提取质量有不利的影响。
实验室间提取方法的可重复性
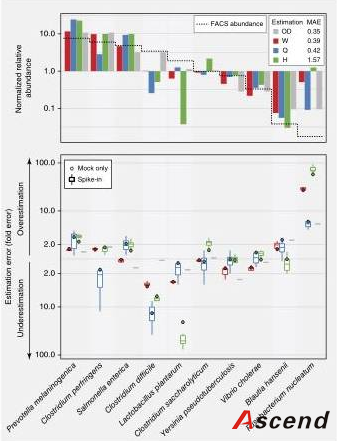
基于对所提取的DNA的质量和菌群多样性的考虑,作者选择了5个最好的方法,分别为方法15、7、6、9和1(按此顺序),来研究实验室间的可重复性(第二阶段)。方法15、6和9使用相同的溶解试剂和试剂盒,因此稍作修改合并成一个方法,作者称之为Q(补充方法),方法1和7分别称之为H和W。最初使用这些方法的实验室在第二阶段中重复使用这些方法,以确保与第一阶段提取产物的差异相当(补充结果Fig. 5)。每种方法都在之前没有经验的实验室同时进行,以评估其做为标准方法的广泛适用性。结果表明,这三种方法在不同的地点都是可重复的,但只有方法H的影响低于最小生物学变异,方法W和Q所产生的变异与样本间的变异相似(补充结果Fig. 5)。
尽管方法H在实验室间有更好的可重复性,但相比另两个方法不适用于革兰氏阳性菌,因其减少了所获得微生物的多样性。方法W也有较高的可重复性,但由于使用了酚氯仿而难以实现自动化。方法Q所得的结果有较高的多样性,似乎能有效溶解革兰氏阳性菌,并容易使用不同的设备来进行操作。
讨论
作者对DNA提取质量进行了比较和重复性分析,其中方法Q似乎是最好的,适合于大多数研究。虽然作者只将方法Q用于测试粪便,但可能也适用于其他样品。实验表明,DNA提取方法的不同对所观察到的微生物组成有极大的影响。实验室间的研究建议应用合适的方法以提高多项研究的可比性。
作者:Costea PI, et al.【European Molecular Biology Laboratory, Germany.】
编译:周志远【辽宁师范大学生命科学学院】
校审:蒲春文【大连市第六人民医院】
DNA提取试剂盒:www.ex-dna.com